Michaelis-Menten kinetics
In biochemistry, Michaelis–Menten kinetics is one of the simplest and best-known models of enzyme kinetics. It is named after German biochemist Leonor Michaelis and Canadian physician Maud Menten. The model takes the form of an equation describing the rate of enzymatic reactions, by relating reaction rate to , the concentration of a substrate S. Its formula is given by
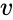 to
to ![{\displaystyle [S]}](https://wikimedia.org/api/rest_v1/media/math/render/png/292bbb82029aa583c5d2ac5fa1d7e4fedf537d8b) , the concentration of a substrate S. Its formula is given by
, the concentration of a substrate S. Its formula is given by
![{\displaystyle v={\frac {d[P]}{dt}}={\frac {V_{\max }{[S]}}{K_{m}+[S]}}.}](https://wikimedia.org/api/rest_v1/media/math/render/png/7a2e1c79e2fc2b111022c4764cee47f4ae18a027)
Here, represents the maximum rate achieved by the system, at maximum (saturating) substrate concentrations. The Michaelis constant is the substrate concentration at which the reaction rate is half of . Biochemical reactions involving a single substrate are often assumed to follow Michaelis–Menten kinetics, without regard to the model's underlying assumptions.
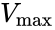 represents the maximum rate achieved by the system, at maximum (saturating) substrate concentrations. The Michaelis constant
represents the maximum rate achieved by the system, at maximum (saturating) substrate concentrations. The Michaelis constant 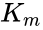 is the substrate concentration at which the reaction rate is half of
is the substrate concentration at which the reaction rate is half of Model
In 1903, French physical chemist Victor Henri found that enzyme reactions were initiated by a bond between the enzyme and the substrate.[1] His work was taken up by German biochemist Leonor Michaelis and Canadian physician Maud Menten who investigated the kinetics of an enzymatic reaction mechanism, invertase, that catalyzes the hydrolysis of sucrose into glucose and fructose. In 1913, they proposed a mathematical model of the reaction.[2] It involves an enzyme E binding to a substrate S to form a complex ES, which in turn is converted into a product P and the enzyme. This may be represented schematically as
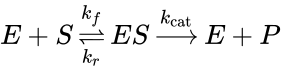
where , and denote the rate constants, and the double arrows between S and ES represent the fact that enzyme-substrate binding is a reversible process.
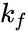 ,
, 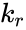 and
and 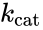 denote the rate constants, and the double arrows between S and ES represent the fact that enzyme-substrate binding is a reversible process.
denote the rate constants, and the double arrows between S and ES represent the fact that enzyme-substrate binding is a reversible process.
Under certain assumptions — such as the enzyme concentration being much less than the substrate concentration — the rate of product formation is given by
![{\displaystyle v={\frac {d[P]}{dt}}=V_{\max }{\frac {[S]}{K_{m}+[S]}}=k_{\mathrm {cat} }[E]_{0}{\frac {[S]}{K_{m}+[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/png/7797c49e7cd63179fcf26d8a596b9489c174da3f)
The reaction rate increases with increasing substrate concentration , asymptotically approaching its maximum rate , attained when all enzyme is bound to substrate. It also follows that , where is the enzyme concentration. , the turnover number, is maximum number of substrate molecules converted to product per enzyme molecule per second.
![{\displaystyle V_{\max }=k_{\mathrm {cat} }[E]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/png/ec8ad5bbfbdd33c4154648155442145135224ca2) , where
, where ![{\displaystyle [E]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/png/409eb69aba4c3afa67a48af6d9f976a28445c544) is the enzyme concentration.
is the enzyme concentration. The Michaelis constant is the substrate concentration at which the reaction rate is at half-maximum, and is an inverse measure of the substrate's affinity for the enzyme. A small indicates high affinity, meaning that the rate will approach more quickly. The value of is dependent on both the enzyme and the substrate, as well as conditions such as temperature and pH.
The model is used in a variety of biochemical situations other than enzyme-substrate interaction, including antigen-antibody binding, DNA-DNA hybridization and protein-protein interaction. It can be used to characterise a generic biochemical reaction, in the same way that the Langmuir equation can be used to model generic adsorption of biomolecular species. When an empirical equation of this form is applied to microbial growth it is sometimes called a Monod equation.
Applications
Parameter values vary wildly between enzymes:
| Enzyme | Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle K_m} (M) | Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle k_\text{cat}} (1/s) | Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle k_\text{cat}/K_m} (1/M.s) |
|---|---|---|---|
| Chymotrypsin | 1.5 × 10−2 | 0.14 | 9.3 |
| Pepsin | 3.0 × 10−4 | 0.50 | 1.7 × 103 |
| Tyrosyl-tRNA synthetase | 9.0 × 10−4 | 7.6 | 8.4 × 103 |
| Ribonuclease | 7.9 × 10−3 | 7.9 × 102 | 1.0 × 105 |
| Carbonic anhydrase | 2.6 × 10−2 | 4.0 × 105 | 1.5 × 107 |
| Fumarase | 5.0 × 10−6 | 8.0 × 102 | 1.6 × 108 |
The constant is a measure of how efficiently an enzyme converts a substrate into product. It has a theoretical upper limit of 108 – 1010 /M.s; enzymes working close to this, such as fumarase, are termed superefficient.
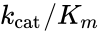 is a measure of how efficiently an enzyme converts a substrate into product. It has a theoretical upper limit of 108 – 1010 /M.s; enzymes working close to this, such as fumarase, are termed superefficient.
is a measure of how efficiently an enzyme converts a substrate into product. It has a theoretical upper limit of 108 – 1010 /M.s; enzymes working close to this, such as fumarase, are termed superefficient.
Michaelis–Menten kinetics have also been applied to a variety of spheres outside of biochemical reactions, including alveolar clearance of dusts, the richness of species pools, clearance of blood alcohol, the photosynthesis-irradiance relationship and bacterial phage infection.
Derivation
Applying the law of mass action, which states that the rate of a reaction is proportional to the product of the concentrations of the reactants, gives a system of four non-linear ordinary differential equations that define the rate of change of reactants with time Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle t} :
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \begin{array}{cccccccc} d [S] / d t & = & - & k_f [E] [S] & + & k_r [ES] & \\ d [E] / d t & = & - & k_f [E] [S] & + & k_r [ES] & + & k_\mathrm{cat} [ES] \\ d [ES] / d t & = & + & k_f [E] [S] & - & k_r [ES] & - & k_\mathrm{cat} [ES] \\ d [P] / d t & = & & & + & k_\mathrm{cat} [ES] \end{array} }
In this mechanism, the enzyme E is a catalyst, which only facilitates the reaction, so its total concentration, free plus combined, Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle [E] + [ES] = [E]_0} is a constant. This conservation law can also be obtained by adding the second and third equations above.
Equilibrium approximation
In their original analysis, Michaelis and Menten assumed that the substrate is in instantaneous chemical equilibrium with the complex, and thus . Combining this relationship with the enzyme conservation law, the concentration of complex is
![{\displaystyle k_{f}[E][S]=k_{r}[ES]}](https://wikimedia.org/api/rest_v1/media/math/render/png/6753d45b4eff88eb9988ecd9397abc6ec205c3f2) . Combining this relationship with the enzyme conservation law, the concentration of complex is
. Combining this relationship with the enzyme conservation law, the concentration of complex is
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle [ES] = \frac{[E]_0 [S]}{K_d + [S]}}
where Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle K_d = k_r / k_f} is the dissociation constant for the enzyme-substrate complex. Hence the velocity Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle v} of the reaction — the rate at which P is formed — is
![{\displaystyle v={\frac {d[P]}{dt}}={\frac {V_{\max }{[S]}}{K_{d}+[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/png/61d1e492354949b99978e2b4ce255a54d3ca9359)
where Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle V_\max = k_\mathrm{cat} [E]_0} is the maximum reaction velocity.
Quasi-steady-state approximation
An alternative analysis of the system was undertaken by British botanist G. E. Briggs and British geneticist J. B. S. Haldane in 1925. They assumed that the concentration of the intermediate complex does not change on the time-scale of product formation — known as the quasi-steady-state assumption or pseudo-steady-state-hypothesis. Mathematically, this assumption means Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle k_f [E] [S] = k_r [ES] + k_\mathrm{cat} [ES] } . Combining this relationship with the enzyme conservation law, the concentration of complex is
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle [ES] = \frac{[E]_0 [S]}{K_m + [S]}}
where
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle K_m = \frac{k_r + k_\mathrm{cat}}{k_f}}
is known as the Michaelis constant. Where Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle k_r} , Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle k_\mathrm{cat}} , and Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle {k_f}} ; are respectively the constants for substrate unbinding, conversion to product, and binding to the enzyme. Hence the velocity Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle v} of the reaction is
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle v = \frac{d [P]}{d t} = \frac{V_\max {[S]}}{K_m + [S]}}
Assumptions and limitations
The first step in the derivation applies the law of mass action, which is reliant on free diffusion. However, in the environment of a living cell where there is a high concentration of proteins, the cytoplasm often behaves more like a gel than a liquid, limiting molecular movements and altering reaction rates. Whilst the law of mass action can be valid in heterogeneous environments, it is more appropriate to model the cytoplasm as a fractal, in order to capture its limited-mobility kinetics.
The resulting reaction rates predicted by the two approaches are similar, with the only difference being that the equilibrium approximation defines the constant as Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle K_d} , whilst the quasi-steady-state approximation uses Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle K_m} . However, each approach is founded upon a different assumption. The Michaelis–Menten equilibrium analysis is valid if the substrate reaches equilibrium on a much faster time-scale than the product is formed or, more precisely, that
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \epsilon_d = \frac{k_\mathrm{cat}}{k_r} \ll 1}
By contrast, the Briggs–Haldane quasi-steady-state analysis is valid if
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \epsilon_m = \frac{[E]_0}{[S]_0 + K_m} \ll 1}
Thus it holds if the enzyme concentration is much less than the substrate concentration. Even if this is not satisfied, the approximation is valid if Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle K_m} is large.
In both the Michaelis–Menten and Briggs–Haldane analyses, the quality of the approximation improves as Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \epsilon\,\!} decreases. However, in model building, Michaelis–Menten kinetics are often invoked without regard to the underlying assumptions.
It is also important to remember that, while irreversibility is a necessary simplification in order to yield a tractable analytic solution, in the general case product formation is not in fact irreversible. The enzyme reaction is more correctly described as
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle E + S \, \overset{k_{f_1}} {\underset{k_{r_1}} {\rightleftharpoons}} \, ES \, \overset{k_{f_2}} {\underset{k_{r_2}} {\rightleftharpoons}} \, E + P }
In general, the assumption of irreversibility is a good one in situations where one of the below is true:
1. The concentration of substrate(s) is very much larger than the concentration of products:
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle [S] \gg [P] }
This is true under standard in vitro assay conditions, and is true for many in vivo biological reactions, particularly where the product is continually removed by a subsequent reaction.
2. The energy released in the reaction is very large, that is
- Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle \Delta{G} \ll 0}
In situations where neither of these two conditions hold (that is, the reaction is low energy and a substantial pool of product(s) exists), the Michaelis–Menten equation breaks down, and more complex modelling approaches explicitly taking the forward and reverse reactions into account must be taken to understand the enzyme biology.
Determination of constants
The typical method for determining the constants Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle V_\max} and Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle K_m} involves running a series of enzyme assays at varying substrate concentrations Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle [S]} , and measuring the initial reaction rate Failed to parse (MathML with SVG or PNG fallback (recommended for modern browsers and accessibility tools): Invalid response ("Math extension cannot connect to Restbase.") from server "https://wikimedia.org/api/rest_v1/":): {\displaystyle v_0} . 'Initial' here is taken to mean that the reaction rate is measured after a relatively short time period, during which it is assumed that the enzyme-substrate complex has formed, but that the substrate concentration held approximately constant, and so the equilibrium or quasi-steady-state approximation remain valid. By plotting reaction rate against concentration, and using nonlinear regression of the Michaelis–Menten equation, the parameters may be obtained.
Before computing facilities to perform nonlinear regression became available, graphical methods involving linearisation of the equation were used. A number of these were proposed, including the Eadie–Hofstee diagram, Hanes–Woolf plot and Lineweaver–Burk plot; of these, the Hanes–Woolf plot is the most accurate. However, while useful for visualization, all three methods distort the error structure of the data and are inferior to nonlinear regression. Nonetheless, their use can still be found in modern literature.
References
This article uses material from the Wikipedia article Michaelis–Menten kinetics, which is released under the Creative Commons Attribution-ShareAlike Unported License 3.0.